析氧反应(OER)是金属空气电池、电解水制氢、燃料电池等新能源技术的关键反应之一。然而,制约其效率的关键在于缓慢的四电子-质子转移过程。在中性电解质中,由于离子浓度低和欧姆损耗大的限制,只有极少数贵金属基水氧化催化剂(WOC)能够有效运行。此外,由于大多数报道的WOCs都是无定形的复合催化剂,没有清晰的晶体结构,因此要在原子水平上深入了解它们的电荷分离过程和催化机理仍然是一个挑战。
自然界中,光合作用系统II(PS-II)的析氧中心(OEC)由Mn4CaO5簇组成,它在中性介质中催化水裂解产生氧气的效率极高。在Mn4CaO5中,相邻的金属中心由μ-oxo桥连接,这被认为是产生OER的关键结构。基于OEC的结构特征,人们设计了各种金属氧化物来模拟PS-II的化学功能。事实上,自然光合作用的主要步骤包括吸收阳光并将其转化为空间上分离的电子/空穴对,而这一步骤经常被忽视。OEC中的空穴能够有效地将水氧化成氧气。因此,在水氧化催化剂(WOCs)中设计一种稳定的电荷分离状态是实现人工光合作用的关键。此外,我们在先前的工作中发现电子转移(ET)光致变色材料具有形成长寿命电荷分离态的潜力。
受到光合作用系统II(PS-II)中析氧中心(OEC)的金属氧簇结构特征和电荷分离行为的启发,我们设计了一种新型晶态光致变色的多钼氧酸盐,MV2[β-Mo8O26](1,MV = 甲基紫精阳离子)。该化合物在光活化后,颜色从无色变为灰色,产生稳定时间长达1年之久的电荷分离态,这种电荷分离态能够有效促进OER活性。着色后,在10 mA cm-2的电流密度下观察到的过电位和塔菲尔斜率分别下降了49 mV和62.8 mV dec-1。着色态在中性电解质中出色的OER性能甚至超过了商用RuO2基准。谱学和理论计算发现光照导致聚阴离子的氧空穴能够促进O-O偶联,增强OER催化活性。
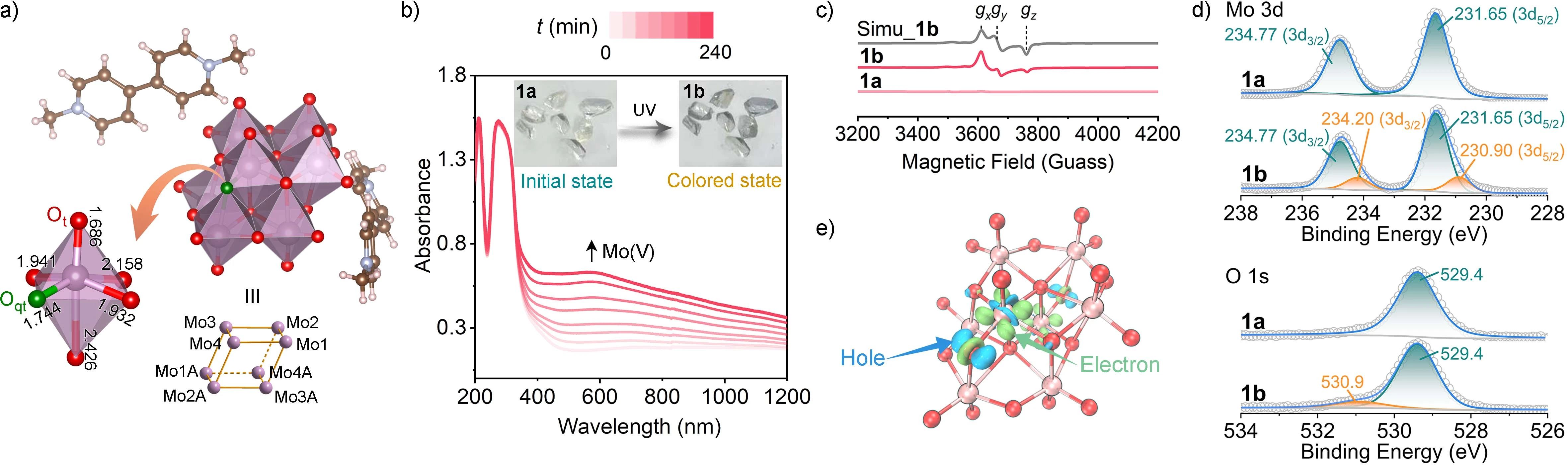
图1. 多钼氧酸盐1。a) 分子结构;b) 时间依赖的紫外-可见吸收光谱,插图:颜色变化;c) 1a、1b和模拟1b的EPR谱;d) Mo 3d和O 1s的XPS芯级谱;e) Mo8的电子密度差,绿色和蓝色分别代表电子转移后的电荷积累和减少,等值面值为0.01 e-Å-3。
多氧钼酸盐(POMo)1由Na2MoO4·2H2O与MVCl2在140℃下的水热反应得到。采用粉末x射线衍射、红外光谱、拉曼光谱和元素分析对1的晶体样品进行相纯度评估。热重分析表明,1至少在350°C以下是热稳定的。1的分子结构一个独立的[β-Mo8O26]4-(Mo8)阴离子和两个MV阳离子。Mo8由8个共边的MoO6八面体构成,形成一个中心对称的平行六面体形状的簇,其中8个Mo原子位于顶点上。值得注意的是,在Mo8簇中,有6个MoO6 (Mo1-Mo3, Mo1A-Mo3A)八面体含有两个末端氧原子(Ot),即每个都含有两个Mo-O短键(<1.75Å)。其余的两个MoO6(Mo4, Mo4A)八面体也都有两个短键,但只有一个末端氧原子,因为其中一个短键上的氧原子是准末端氧原子(表示为Oqt,如图1a中绿色所示),它在两个Mo原子之间起μ2-O的作用。
通过紫外光辐照,多钼氧酸盐1由无色(1a)变为灰色(1b)。电子顺磁共振(EPR)证实了辐照后MoV的出现和光诱导电子转移的发生。X射线光电子能谱(XPS)进一步表明Mo是电子受体,O原子为电子给体。电子密度差(Δρ)计算结果展示了在光照下电子从准端氧(Oqt)转移到相邻的Mo,在Oqt留下空穴,从而形成超稳定的电荷分离态。计算的ADCH电荷和Hirshfeld电荷表明,与初始状态相比,电荷分离态的Oqt具有更正的电荷,而相邻的Mo原子表现出更负的电荷,这进一步证明了辐照后电子从Oqt转移到相邻的Mo。

图2. OER的电催化性能。a) 样品1a、1b、2、3a、3b和商用RuO2在氧气饱和的0.1 M PBS溶液中的LSV曲线,扫描速率为5 mV s-1;b) 10 mA cm-2电流密度下的过电位;c) Tafel斜率;d) 在1.60 V(RHE)的外加电压下获得的奈奎斯特图与和对应等效电路图(插图)。
PM6-D3H4计算表明了这种超稳定的电荷分离态是源于紫精π-阳离子对Mo8强的极化作用。这个结论可以进一步通过分析两种设计的多钼氧酸盐【TEA2[β-Mo8O26](H3O)2 (2, TEA = 四乙基胺阳离子) 和and PD4[β-Mo8O26](H2O)4 (3, PD = 哌啶鎓阳离子)】概念验证模型所证实。
样品在中性电解质中OER的性能结果表明光活化可以有效降低过电势。在10 mA cm-2的电流密度下观察到1b的过电位和Tafel斜率分别下降了49 mV和62.8 mV dec-1。1b代表了目前中性介质中OER性能最好的非贵金属催化剂之一。此外,通过对比另外两种多钼氧酸盐2和3的OER性能,表明了1中的MV的存在可以作为电子转移中继物来有效促进电荷传输。
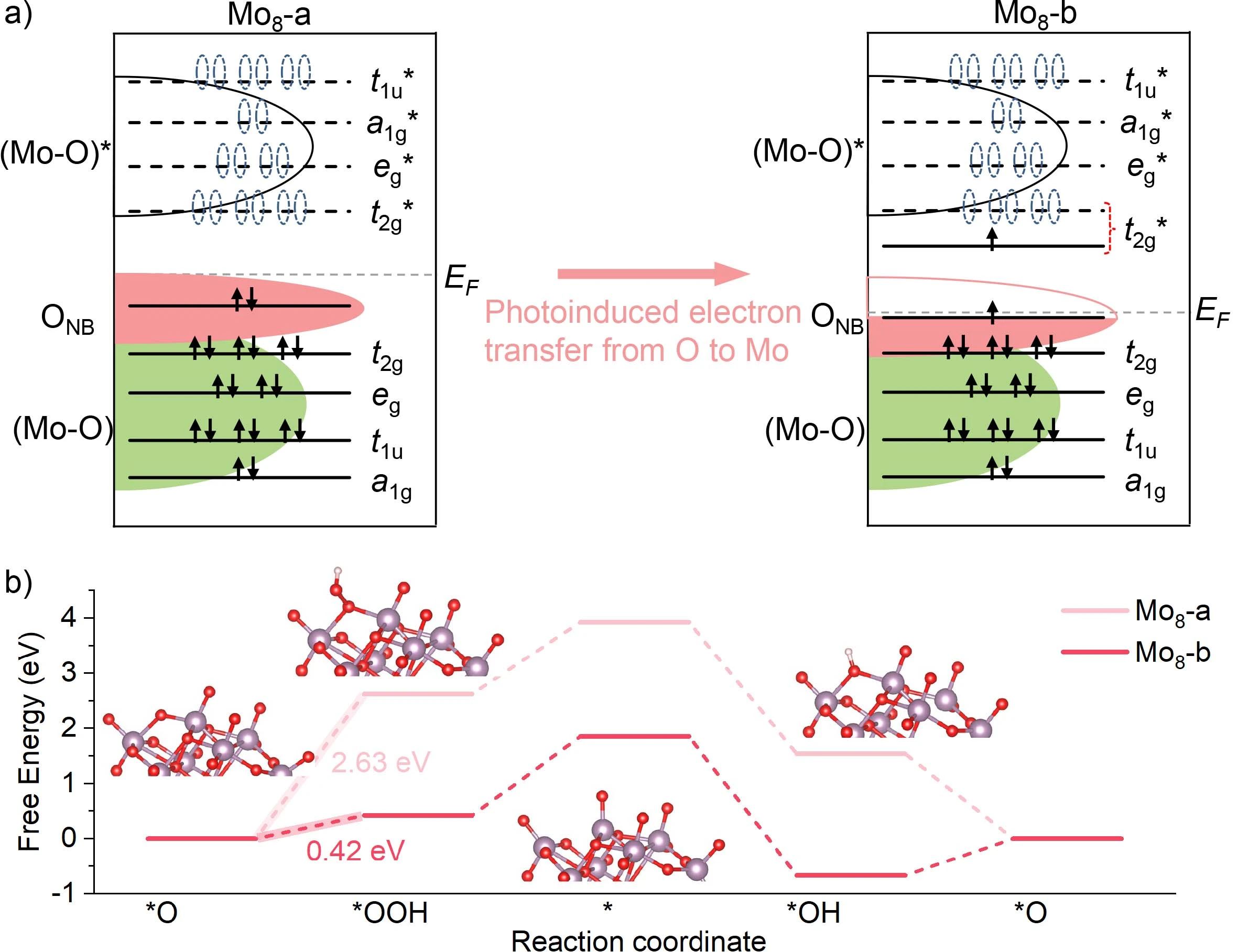
图3. 辐照后Mo8从初始状态到电荷分离状态的电子构型变化示意图,ONB表示氧中的非键电子,EF表示费米级;b) 在1.23 V的外加电位下,Mo8的OER阶自由能。
最后,DFT计算进一步研究了OER机制。能带结构和电子构型分析表明,Oqt的非键电子转移到Mo后,费米能级降低,表明Oqt上空穴的氧化能力增强。通过计算得到的Mayer键级分析可知,在电子转移后,Mo8-b的Mo-Oqt键级较初始状态明显降低,表明Mo-Oqt键被削弱和激活。因此,选择Oqt原子作为吸附中间产物的活性位点来计算自由能图和反应路径。反应从形成O-O键生成*OOH开始,然后解吸生成氧气,再吸附OH-离子,最后去质子化生成*O。在U=1.23V时,Mo8-a的速率决定步骤(RDS)是从O*到OOH*形成O-O键。Mo8-a的RDS能垒为2.63 eV,而Mo8-b的能垒为0.42 eV,这表明辐照后Oqt上的空位能很好地促进O-O直接偶联。
总的来说,研究者提出的光致变色增益OER活性的策略为今后开发用于人工光合作用装置的非贵金属基OER催化剂提供了一个前瞻性的方向和设计思路。
成果以“Photo-Activating Biomimetic Polyoxomolybdate for Boosting Oxygen Evolution in Neutral Electrolytes”为题发表在Angew. Chem. Int. Ed.上,betway必威官网博士研究生李大欢和硕士生张潇月为共同第一作者,孙财副教授和郑寿添教授为通讯作者。